In this article:
- Why we developed a better specificity testing tool
- What advantages does the MPA offer over conventional methods?
- What is the ISTAND qualification process?
- What makes the MPA’s qualification significant?
This article is part of a series about specificity testing. Be sure to read part 3, about the limitations of tissue cross-reactivity studies.
Summary
Tissue cross-reactivity (TCR) studies have served as the standard for specificity testing since the 1980s, but their limitations—inability to identify specific proteins, subjective interpretation, poor clinical correlation—have long been recognized. The Membrane Proteome Array (MPA) was designed to address these gaps with objective, quantitative data that identifies specific off-target proteins. In 2021, we began working with the FDA to qualify the MPA as a Drug Development Tool through the ISTAND program. While the FDA already accepts MPA data, this qualification process validates the scientific rigor behind the platform and helps formalize the shift toward more predictive, human-relevant specificity testing methods.
Why we built a better specificity testing tool
As discussed in the previous article, tissue cross-reactivity (TCR) studies leave significant gaps in our understanding of therapeutic specificity. Most critically, they can’t tell you which protein a therapeutic is binding to—only where unexpected staining appears in tissues. That makes it nearly impossible to assess the actual safety risk or design appropriate follow-up studies.
Drug developers and regulators have long recognized the need for better tools. The FDA stated as far back as 1997 that “appropriate newer technologies should be employed as they become available and validated.” More recently, their 2024 CAR-T guidance specifically named “protein arrays” as an acceptable alternative to TCR.
The Membrane Proteome Array (MPA) was designed to fill the gaps left by TCR. And the shift in the regulatory landscape has already begun: MPA data has already been accepted in over 100 IND applications. Proper specificity assessment early in development contributes to better candidate selection, more efficient regulatory review, safer clinical trials, and ultimately better therapeutics for patients.
What advantages does the MPA have over conventional methods?
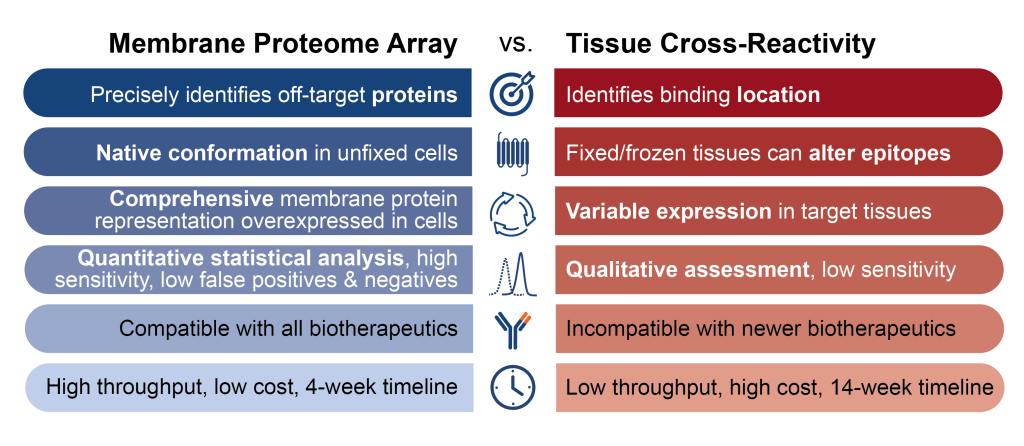
The MPA offers several key advantages over conventional specificity testing methods:
-
-
- Identifies specific proteins. When the MPA detects off-target binding, it tells you exactly which proteins are involved. That enables focused investigation into potential safety issues and informed decisions about whether to proceed with development. TCR studies, by contrast, can only show you tissue locations.
- Uses native protein conformations. Each protein is expressed in its natural state within whole eukaryotic cells, with proper folding, post-translational modifications, and membrane environment. TCR tissue processing can alter protein structures, potentially causing false positives or negatives.
- Eliminates donor variability. The MPA uses established cell lines with consistent handling protocols, so every protein is fully expressed and available for testing. TCR’s reliance on tissue samples from three individual donors means protein expression varies unpredictably.
- Provides objective, quantitative data. The MPA uses flow cytometry to generate measurements that can be statistically analyzed and compared across studies. This eliminates the subjectivity inherent in TCR, where pathologists score immunohistochemistry staining patterns.
-
Additionally, MPA data supports regulatory submissions directly. MPA results provide the precise, quantitative data needed for IND applications. For candidates without off-targets, the specificity is demonstrated clearly. For those with off-targets, you have the molecular detail needed to design appropriate follow-up studies.
These advantages made the MPA an ideal candidate for FDA qualification as a Drug Development Tool—a formal recognition that would help accelerate the broader shift toward more predictive specificity testing methods.
What is the ISTAND qualification process?
In May 2021, Integral Molecular submitted a Letter of Intent to the FDA’s ISTAND program to qualify the MPA as a Drug Development Tool. ISTAND (Innovative Science and Technology Approaches for New Drugs) provides a pathway for qualifying novel methods that can improve drug development and regulatory review.
The FDA accepted the MPA into ISTAND in July 2022, making it the first tool ever accepted into the program. This milestone reflected both the platform’s scientific merit and the FDA’s recognition that better specificity testing methods are needed.
The qualification process involves several stages:
-
-
- Letter of Intent (LOI) - Describes the tool, its intended use, and preliminary data supporting its utility. Accepted July 2022.
- Qualification Plan (QP) - Outlines the validation studies, performance characteristics, and regulatory strategy. Submitted August 2023, accepted January 2025.
- Full Qualification Package (FQP) - Provides comprehensive validation data and evidence supporting the tool’s use in regulatory submissions. Submitted Q4 2025.
-
Throughout this process, the FDA has provided feedback and suggestions for enhancements. Their input has helped transform an already strong platform into one that’s even more robust, reproducible, scalable, and well-documented.

What makes the MPA’s qualification significant?
While the FDA already accepts MPA data, qualification will streamline the regulatory review process. Once the FDA qualifies the MPA, any drug developer can use it in their IND applications “with confidence that the FDA will accept the data” (Roadmap to Reducing Animal Testing in Preclinical Safety Studies).
The MPA will likely represent several firsts in the New Approach Methodology (NAM) and Drug Development Tool landscape:
-
-
- The only NAM specificity test with published validation data
- The first qualified NAM for specificity testing
- On track to be the first NAM qualified through the ISTAND process
- On track to be the first NAM qualified as a DDT by the FDA
-
For organizations considering qualifying their own drug development tools through ISTAND, we highly recommend it. The experience has been invaluable, and the FDA’s input has genuinely enhanced the platform.
Looking ahead
In the coming articles, we’ll share more details about how the MPA works, including the proteins represented in the library, the screening process, and the quality systems that ensure reliable results. We’ll also discuss how MPA data compares to TCR studies and share insights into minimizing false positives and false negatives.